INTRODUCTION
Sickle cell disease (SCD) is an autosomal recessive condition that results from the presence of a mutated form of hemoglobin. It is the most common inherited blood disorder in the United Kingdom, where it affects 12,000ŌĆō15,000 people among approximately 250,000 carriers of the sickle cell gene [1]. Globally, SCD is most common among people of West African or Caribbean descent [2]. The cause of SCD is a single nucleotide substitution in the beta globin gene on the short arm of chromosome 11. The resulting protein, hemoglobin S, polymerizes under low oxygen conditions, causing red blood cells to become rigid and sickle-shaped. These sickle cells cause the activation of other circulating cells, including neutrophils, and blood vessel walls as part of processes similar to those observed in vasculopathies. The tissue subsequently loses its blood supply, and the resulting ischemia causes tissue infarction [3]. The most common manifestations are painful vasoocclusive crisis (VOC), hemolytic anemia, and end organ damage caused by vasculopathy and tissue ischemia. Complications may involve a sudden onset, which is known as a sickle cell crisis, although some degree of sickling typically also leads to long-term organ damage. The VOC results from obstruction of the microvasculature by sickled red blood cells, which causes ischemia and pain. Crises may occur for no apparent reason, or there may be triggering a factor. Common triggering factors include dehydration, a sudden change in body temperature (e.g., caused by infection or environmental factors), or hypoxia caused by stress or exercise [4]. Fetal hemoglobin (HbF) levels are highly variable and inheritable, primarily through genetic variants at three principal loci: BCL11A, HBS1L-MYB, and the HBB cluster, which account for 10%ŌĆō20% of HbF variations among SCD patients [5,6]. Some variations in BCL11A are amenable to therapeutic manipulation, which led to the reversal of SCD symptoms in mouse models [7]. Functional studies have shown that the expression of BCL11A (a transcriptional repressor) is regulated by erythroid-specific enhancers that contain 3ŌĆÖ DNase hypersensitive sites (DHS) located +62, + 58, and +55 kb from the transcription initiation site of BCL11A [8]. Two single-nucleotide polymorphisms (SNPs) in the enhancer element have a strong association with HbF levels among African American SCD patients: rs1427407 in DHS +62 and rs7606173 in DHS +55. This is consistent with the hypothesis that multiple functional SNPs act in combination to influence BCL11A regulation [5,8,9]. The present study investigated the relationship between a variant of BCL11A (rs1427407) and its plasma levels during VOC and stroke complications among SCD patients from Khartoum state, Sudan. The findings of this study may improve our ability to judge the severity of the sickle cell anemia (SCA), which can in turn help to improve its clinical management.
METHODS
This cross-sectional study was performed between June 2014 and October 2016 in the Omdurman State Teaching Hospital, Sudan. The study included 166 patients who were diagnosed with SCD or been hospitalized for treatment of painful VOC during the 6 months before the study. The analysis of VOC was restricted to subjects with a confirmed diagnosis of SCA. The patients were grouped according to sex and age group (<15 years, 15ŌĆō25 years, and >25 years). In addition, 35 healthy subjects were recruited to create a control group with similar age and sex characteristics. The subjects and/or their guardians provided informed consent to participate in the study after receiving a detailed explanation regarding the purpose of the study and any related risks. Blood samples were collected from the patients and controls under aseptic conditions using an EDTA-coated vacutainer tube.
A complete blood count was performed using a hematology analyzer (Sysmex XP-300/XP-100, Sysmex Asia Pacific Pte Ltd, Jalan Tukang, Singapore). Levels of HbF were measured using high-performance liquid chromatography (BIO-RAD, Hercules, CA, USA). Plasma BCL11A levels were determined using an enzyme-linked immunosorbent assay (FY0208A; Hu Feng Biological Technology, Shanghai, China). The patientsŌĆÖ peripheral blood samples were used to collect DNA samples according to the manufacturerŌĆÖs instructions (Puregene blood package; Qiagen, Germantown, MD, USA). Molecular analysis to detect the sickle mutation was performed using 200 ng DNA and polymerase chain reaction (PCR) amplification of the ╬▓-globin gene with the appropriate oligonucleotide primers (sense: 5ŌĆÖ-CACTGAACCCCCCACCTACCA-3ŌĆÖ, antisense: 5ŌĆÖ-CTCCACTCCCCGTACCTTCC-3ŌĆÖ). Direct sequencing was performed using the ABI prism 3730XL DNA sequencer (Applied Biosystems, Foster City, CA, USA), based on DDE I digestion of the PCR product [5,10]. Multiplex snap PCR and capillary electrophoresis were used to determine the rs1427407 genotype [11].
All statistical analyses were performed using IBM SPSS ver. 19.0 (IBM Corp., Armonk, NY, USA). Results were expressed as mean┬▒standard deviation and were compared between the patients and controls using the t-test or FisherŌĆÖs exact test. Differences were considered statistically significant at P-values of <0.05. The allele frequency was calculated based on the different genotype distributions.
RESULTS
The characteristics and laboratory test results for the SCD patients and controls are shown in Table 1. There were no significant differences between the two groups according to age (P=0.9239) or sex (P=0.4316). In addition, no significant differences were observed between the 116 SCD patients and the 35 controls according to mean red blood cell count (P=0.3916), mean corpuscular volume (P=0.9426), or mean corpuscular hemoglobin level (P=0.5288). However, there were significant differences in the mean white blood cell count (P=0.0001), platelet count (P=0.0001), HbF percentage (P=0.0001), reticulocyte count (P=0.0001), and plasma BCL11A level (P=0.0001).
The rs1427407 sequencing revealed three genotypes: GG, GT, and TT. The distributions of the genotypes among the SCD patients and healthy controls are shown in Table 2. Our results have been verified by the fact that SCD is associated with the BCL11A genotype polymorphism. The BCL11A genotypes that included the T allele and the G allele at rs1427407 were common among SCD patients. We evaluated whether SCD complications were associated with the various genotypes, and found that the BCL11A rs1427407 GG and GT genotypes had significantly elevated risks of SCD complications (P=0.0008) (Table 2).
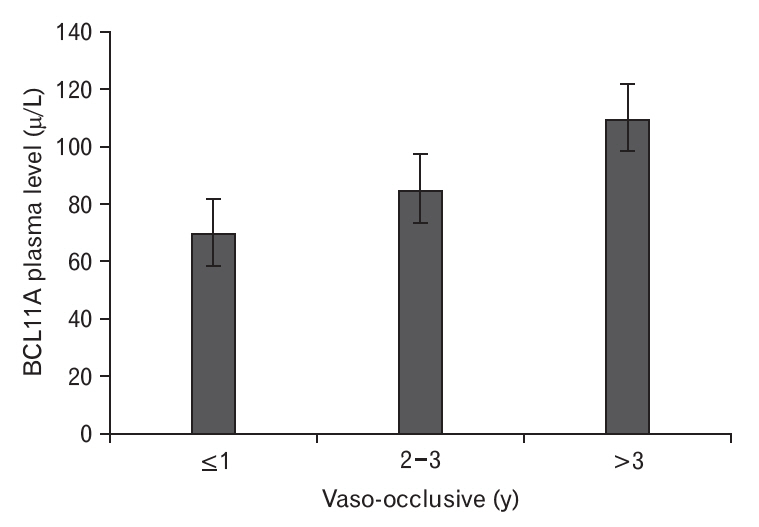
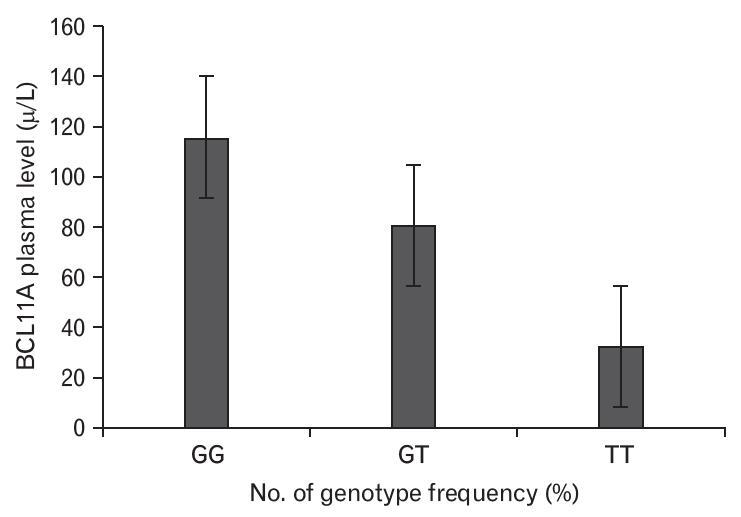
The plasma BCL11A levels were measured in SCD patients according to their VOC duration (Figure 1), which revealed that plasma BCL11A levels were elevated among patients with a VOC duration of >3 years. In addition, plasma BCL11A levels were higher in patients with the GG genotype than in patients with the GT and TT genotypes (Figure 2). The frequencies of the BCL11A rs1427407 genotypes according to SCD complications are shown in Table 3. We found that the GG, GT, and TT genotypes had significantly different VOC durations (P=0.0004) and significantly different rates of stroke (P=0.008) (Table 3). Thus, the BCL11A rs1427407 GG/GT genotypes appear to be associated with increased risks of VOC and stroke among SCD patients.
DISCUSSION
The BCL11A rs1427407 variant is commonly found in SCD patients [12]. The BCL11A gene was first detected in B-cell chronic lymphocytic leukemia and is a protooncogene for malignant hematological diseases [13,14]. Our study revealed that SCD patients had plasma BCL11A levels that were significantly higher than the control subjects (100.13 ╬╝g/L versus 70.88 ╬╝g/L, P<0.05). Furthermore, the SCD patients had a higher frequency of the GG and GT genotypes at BCL11A rs1427407. Moreover, the BCL11A rs1427407 GG variant increased the risk of developing SCD (Table 2). Some studies have also indicated that BCL11A can be used to predict overall and disease-free survivals [15,16]. In our study, the plasma BCL11A levels increased according to the duration of VOC in SCA patients. However, previous studies have revealed conflicting data regarding the associations between BCL11A variants and SCA-related complications, such as VOC frequency [11,17] and stroke [11,18]. We observed statistically significant problems with the inheritance of BCL11A rs1427407 in >3 VOC/year or stroke, while the frequency of BCL11A rs1427407 genotypes was significantly related to vaso occlusive (VOc) periods in SCA patients (Table 3). Therefore, plasma BCL11A levels appear to be higher for more advanced VOC relative to the more initial stages (Figure 1). Our study also revealed that the BCL11A rs1427407 genotypes (GG/GT/TT) were significantly associated with stroke complications among SCD patients, and this finding agrees with the results of Satraf et al. [19] The presence of BCL11A rs1427407 appears to reflect a genetic hazard profile with a higher reticulocyte percentage and higher risk of stroke (Table 3). The association with VOC duration of >3 years also indicates that this genetic profile was strongly associated with markers of hemolysis and it appears that the presence of SCA is related to stroke complications. Our findings also agree with a previous report that a decreased hemoglobin level is a risk factor for stroke (Table 1) [19], and that increased markers of hemolysis are independently associated with increased reticulocytes and silent strokes [20]. Relative to the studies performed by Saraf et al. [19] and Kwiatkowski et al. [21], our study has several useful parameters. For example, regardless of the cross-sectional design, we evaluated variations in stroke history according to the BCL11A rs1427407 genotypes by retrospectively collecting stroke records that were prepared using a medical evaluation tool. This may have reduced the risk of detection bias and protected against including silent infarcts in this analysis, which increased the likelihood of identifying VOC and stroke in SCD patients.
In conclusion, the present study revealed that the BCL11A rs1427407 genotypes and elevated BCL11A plasma levels were associated with the risk of stroke, which indicates that genetic factors influence the degree of hemolysis and HbF levels, which may improve our ability to identify SCA patients who are experiencing these complications. Furthermore, plasma BCL11A levels were elevated in patients with the GG/GT genotype of BCL11A rs1427407. To the best of our knowledge, this is the first study to evaluate the genetic influence on SCA severity among Sudanese patients, which may help improve the medical management of these cases. However, our findings must be validated by larger studies. Nevertheless, if further research confirms our results, our findings may indicate that genetic evaluations can help improve the treatment of patients with SCA who are experiencing stroke complications or VOC.