A Diagnosis to Consider in an Adult Patient with Facial Features and Intellectual Disability: Williams Syndrome
Article information
Abstract
Williams syndrome (OMIM #194050) is a rare, well-recognized, multisystemic genetic condition affecting approximately 1/7,500 individuals. There are no marked regional differences in the incidence of Williams syndrome. The syndrome is caused by a hemizygous deletion of approximately 28 genes, including ELN on chromosome 7q11.2. Prenatal-onset growth retardation, distinct facial appearance, cardiovascular abnormalities, and unique hypersocial behavior are among the most common clinical features. Here, we report the case of a patient referred to us with distinct facial features and intellectual disability, who was diagnosed with Williams syndrome at the age of 37 years. Our aim is to increase awareness regarding the diagnostic features and complications of this recognizable syndrome among adult health care providers. Williams syndrome is usually diagnosed during infancy or childhood, but in the absence of classical findings, such as cardiovascular anomalies, hypercalcemia, and cognitive impairment, the diagnosis could be delayed. Due to the multisystemic and progressive nature of the syndrome, accurate diagnosis is critical for appropriate care and screening for the associated morbidities that may affect the patient's health and well-being.
INTRODUCTION
Williams syndrome (OMIM #194050) is a rare, well-documented, multisystem genetic condition affecting approximately 1/7,500 individuals. There are no marked regional differences in the prevalence of this disease.1) The syndrome is caused by the hemizygous deletion of approximately 28 genes, including ELN on chromosome 7q11.2.2) Prenatal and postnatal growth retardation, distinctive facial features, cardiovascular abnormalities, and a unique behavioral phenotype are among the most common clinical features of the syndrome.3) Periorbital fullness, a flat malar region, a full nasal tip, a wide mouth, and full lips and cheeks, which are together described as ‘elfin face,’ and stellate irises, especially in green- or blue-eyed individuals, are among the major facial features of patients with Williams syndrome. The facial features of patients with the syndrome do not differ with ethnic background. The exception is the presence of stellate irises, which are rarely reported in Asian patients.4) Supravalvular aortic stenosis is the most common cardiovascular system (CVS) abnormality, followed by supravalvular pulmonary stenosis and peripheral pulmonary stenosis. Mitral valve prolapse (MVP) and regurgitation may also be observed.3) Hypercalcemia is the most common endocrine problem, followed by hypothyroidism, impaired glucose tolerance, and osteopenia.35) Chronic abdominal pain, constipation, and diverticulosis attributed to elastin haploinsufficiency are also seen in patients with Williams syndrome.6) Moderate cognitive impairment and unique hypersocial behavioral features, such as diminished fear to approach strangers, elevated emphatic concern, reduced social fear, an increased attraction towards faces and eyes in social interactions, and a greater interest in contact with adults rather than younger peers are characteristic neurobehavioral features of the syndrome.6) Generalized anxiety disorder or specific phobias may also coexist with the above behavioral characteristics.7) Williams syndrome is usually diagnosed during infancy or childhood. However, in the absence of classical findings, the diagnosis may be challenging. Due to the multisystemic and progressive nature of the syndrome, accurate diagnosis is important for proper management and follow-up of the patients. Here, we report the case of a 37-year-old female patient referred to us with distinct facial features and intellectual disability. Our aim is to thoroughly describe the diagnostic features and management guidelines for Williams syndrome along with its associated complications.
CASE REPORT
A 37-year-old female patient was referred to Hacettepe University, Pediatric Genetics Department following complaints of atypical facial features. She was the second child of non-consanguineous healthy parents. She was born at term by spontaneous vaginal delivery with a birthweight of 1,500 g (lower than 3rd centile). Her birth length and head circumference were not recorded. The patient had delayed psychomotor development and began to walk and speak after the age of 3 years. She has been able to read and write after attending a rehabilitation program. She was evaluated for developmental delay, short stature, learning disability, recurrent upper respiratory system infections, and constipation in childhood, but no diagnosis was made. Laboratory work-up, including cranial imaging, echocardiography, and blood biochemistry were all normal except for subclinical hypothyroidism and osteoporosis. The patient had generalized anxiety disorder and hypochondriasis since her adolescence and her symptoms had deteriorated over time. She started to suffer from difficulty falling asleep, especially during the last five years. She was described as over-friendly by her family members. On her admission, a physical examination revealed a weight of 42 kg (lower than 3rd centile), a height of 145 cm (lower than 3rd centile), and a head circumference of 52 cm (−2 standard deviations). Among the facial features, a long face, a long neck accentuated by sloping shoulders, low-set ears, periorbital fullness, a short nose, a wide mouth, a small jaw, and malar flattening were noted (Figure 1). She was talkative and gregarious. The patient exhibited prolonged phases of staring into our faces and maintained eye contact throughout the physical examination. Based on these findings, the patient was clinically diagnosed with Williams syndrome. Fluorescence in situ hybridization analysis revealed 46,XX.ish del(7)(q11.2 q11.2) (ELN-), which confirmed the clinical diagnosis (Figure 2). Genetic counseling was provided to the family and a follow-up plan was created. An informed consent was obtained from the patient.
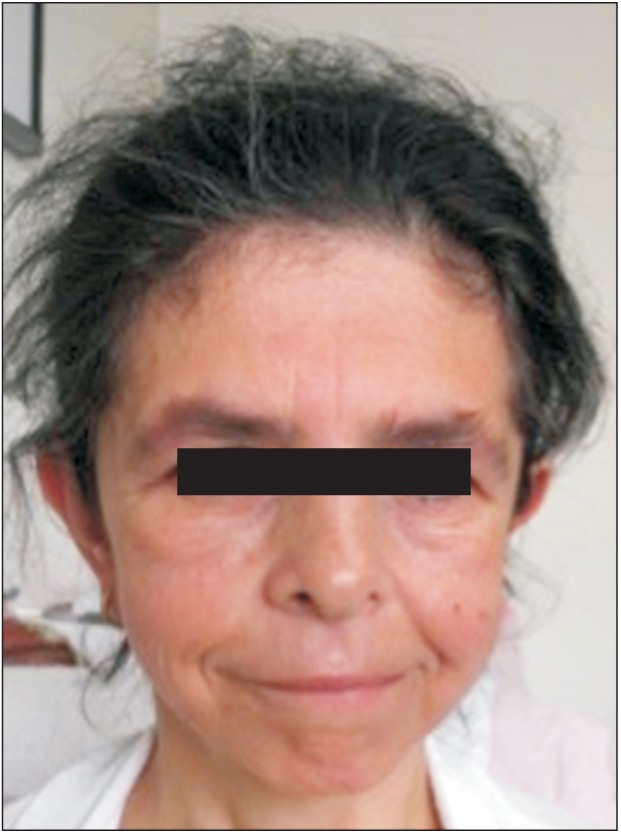
Front view of the patient. Note the long face, periorbital fullness, short nose, wide mouth, small jaw, and malar flattening.
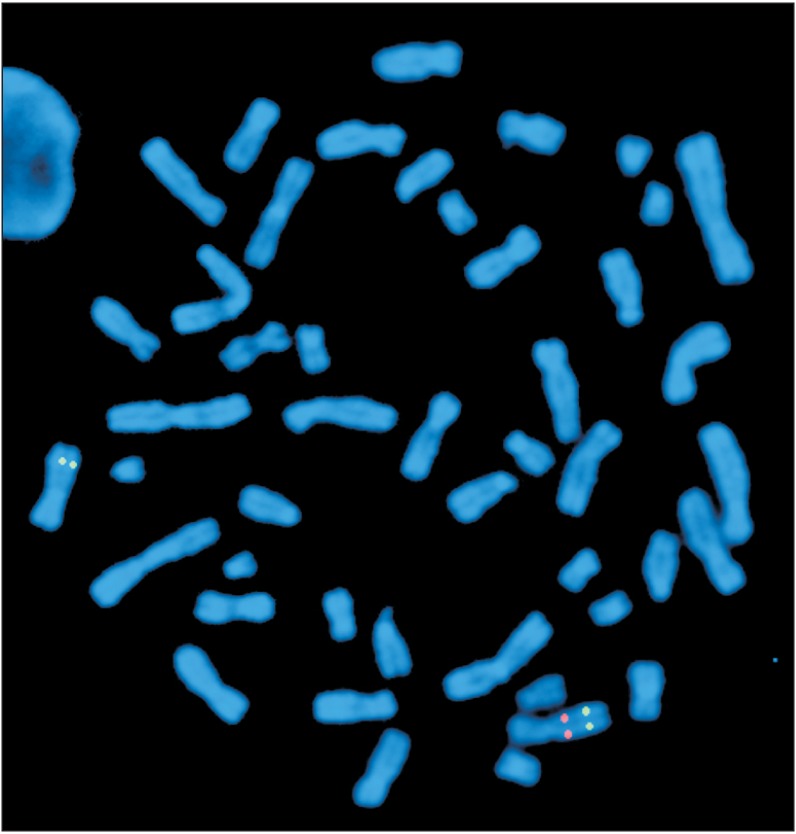
Fluorescent in situ hybridization metaphase image with elastin gene probes. Please notice the absence of the red signal in one copy of chromosome 7, confirming the deletion of 7q11.23.
DISCUSSION
Williams syndrome is a multi-systemic disorder with a wide range of clinical symptoms and associated morbidities. This syndrome is usually diagnosed during infancy or early childhood. The symptoms in adulthood are not as suggestive as they are during childhood. In adulthood, patients usually present with mild intellectual disability and psychiatric disorders, including depression and anxiety disorder.7) The patients usually have subtle findings, but their unique behavioral patterns may be suggestive of Williams syndrome. Therefore, the awareness and shrewdness of the clinician may play a major role in the diagnosis of adult patients. During adulthood, due to the progressive and multi-systemic nature of the syndrome, patients suffer from problems concerning many organ systems and usually receive problem-specific care. With this kind of approach, patients cannot be evaluated in a holistic manner that would lead to the realization that each medical problem is an element of the underlying disorder. Therefore, the diagnosis of Williams syndrome may be challenging. The patient described here was not diagnosed during her infancy or childhood. She was repeatedly evaluated for developmental delay, short stature, and learning disability during childhood. However, no solid diagnosis was reached. She was referred to our clinic with the complaints of atypical facial features and intellectual disability. The quite unique behavioral phenotype was still not pointed out to us upon admission.
The clinical features of Williams syndrome may differ owing to the size of the deleted region.3) During the diagnosis, characteristic facial features and behavioral and cognitive phenotypes are the most helpful components. The distinctive facial appearance may change during adulthood. The fullness of the cheeks resolves, the face appears to be gaunt, and the patient has sloping shoulders and a long neck. Full lips and a wide mouth appear to be the only recognizable facial findings. Patients look older than they are and have premature graying of hair and dental malocclusion.6) In his case, the patient had full lips, a wide mouth, and periorbital fullness, which were key findings in the diagnosis.
The behavioral profile of Williams syndrome is as distinctive as the facial features associated with this condition. Patients have a unique behavioral pattern characterized by hypersociability, heightened empathy, and a desire for close relationships. The behavior of patients with Williams syndrome is characterized by approachability towards strangers and an increased attraction towards faces and eyes. These patients can easily start a conversation, but have difficulties in maintaining it. In contrast to their over-friendly phenotype, patients with Williams syndrome cannot form peer friendships due to difficulties in social adjustment and disinhibition. Social isolation especially manifests itself during adolescence and adulthood. The patient in this case was described by her family members as talkative, friendly, endearing, and gullible, especially during childhood. Her parents reported that she has a tendency to approach strangers and to put herself in danger. Despite her cheerful nature, the patient had generalized anxiety disorder and spells of panic beginning in adolescence. She was always nervous about being lonely and she had fear of illness and death. In Williams syndrome, specific phobias are mainly seen during childhood, while generalized anxiety disorder is the most frequent psychiatric diagnosis in adolescents and adults.7) Diagnosis of anxiety disorders tend to increase with age and significantly affects the quality of life of the patient. Therefore, a low threshold for psychiatric intervention is recommended.6) The patient in this case was diagnosed with generalized anxiety disorder and hypochondriasis during her follow-up.
The cognitive profile of Williams syndrome is also unique and varies from severe mental insufficiency to normal IQ. Patients have profound weaknesses in visuo-spatial constructive ability, but have strengths in language with excellent vocabularies.37) Adaptive functions of the individuals are also reported to be lower than normal. Developmental delay and mild intellectual disability were detected during childhood in the present patient. Regression in mental status was not detected, as expected. The patient lives with her family and takes care of her basic needs, but has never been employed.
Williams syndrome is a progressive disease with multi-systemic complications. The prevalence of these complications may increase with age. CVS, endocrine, and gastrointestinal system problems are frequently reported among adult patients.689) Supravalvular aortic stenosis is the major diagnostic cardiovascular system finding during childhood. However, in adulthood, mitral valve prolapse and regurgitation are the most commonly seen structural cardiac abnormalities. Although our patient did not have any symptoms related to CVS, her evaluation after the establishment of the diagnosis revealed MVP and mild hypertension. Annual CVS follow-ups were planned, as recommended. 36) Hypercalcemia, abnormal glucose intolerance, diabetes mellitus, hypothyroidism, and diminished bone density are the leading endocrine problems in patients with Williams syndrome.5) In our patient, subclinical hypothyroidism and osteoporosis were detected before her referral to our center. However, laboratory results related to hypercalcemia and hyperglycemia were all normal. Routine follow-ups for thyroid function tests every 3 years and annual dual-energy X-ray absorptiometry scans were planned. Gastrointestinal problems are frequently described in the literature, especially in adult patients.6) Chronic constipation, diverticulosis attributed to elastin haplosufficieny, and chronic abdominal pain are the leading problems.6) Our patient had chronic constipation, abdominal pain, and intermittent rectal bleeding. Colonoscopy and rectoscopy revealed multiple rectal fissures and dietary manipulation was recommended.68) Recurrent urinary tract infections, inguinal hernias, and bladder diverticula are common genitourinary system problems in adult patients, among which only bladder diverticulosis was detected in our patient.310)
In conclusion, Williams syndrome is a recognizable syndrome with distinct facial features, structural abnormalities, and unique a behavioral phenotype. However, when these findings are not recognized, diagnosis of the syndrome may be delayed. Diagnosis of Williams syndrome is critical for appropriate patient care and screening for high-risk problems that may affect the patient's health status and well-being. Therefore, the clinician's knowledge of this syndrome and skepticism are the key elements for proper diagnosis. We report this case to increase awareness of Williams syndrome among adult health care providers.
Notes
CONFLICT OF INTEREST: No potential conflict of interest relevant to this article was reported.