INTRODUCTION
Pancytopenia is a condition that can prove to be a diagnostic challenge for primary care physicians. It can be caused by a wide variety of etiologies, namely bone marrow failure, marrow infiltrating lesions, autoimmune disorders, infections, and nutritional deficiencies [1], and is usually managed by a hematologist or a rheumatologist, depending on the etiology. As the potential causes are directly related to various anomalies of the bone marrow, a bone marrow biopsy is one of the most critical investigations for managing a patient with pancytopenia [1,2]. In Korea, malignancy is a common diagnosis from bone marrow biopsies, but a substantial percentage of patients have unspecific findings [2]. Recent terminology describes this condition as idiopathic cytopenia of undetermined significance (ICUS) [3]. However, little is reported regarding the epidemiology and clinical characteristics of this condition. Once autoimmune causes are also ruled out, the affected patients are in what is described as a clinical ŌĆśgray zoneŌĆÖ where neither hematologic nor rheumatologic management alone can completely provide patients with comprehensive medical care. This adversely affects their quality of life as well as clinical status. This case report describes the clinical history and management of a 69-year-old man with longstanding pancytopenia along with splenomegaly and neutropenia, in the department of family medicine at a Korean tertiary hospital. Accordingly, we have explored the role of family medicine in the management of such ŌĆśgray zoneŌĆÖ patients.
CASE REPORT
A 69-year-old man was referred to the department of family medicine at Severance Hospital for chronic weakness and recurrent admissions for untreated and undiagnosed longstanding pancytopenia, which had persisted for the last 10 years. The patient had severe splenomegaly and was being treated for fungal pneumonia (aspergilloma and Pneumocystis jirovecii pneumonia) before the referral. There was past medical history of right thyroidectomy (for thyroid cancer), posterior right coronary artery stenting (due to chronic stable angina pectoris), and paroxysmal atrial fibrillation. The repeated admissions were severely crippling the patientŌĆÖs quality of life. The current admission was for exacerbation of fungal pneumonia. Upon initial evaluation, the patient was afebrile, with body temperature of 36.5┬░C and blood pressure of 99/66 mm Hg. Heart rate was 77 beats per minute and respiratory rate was 20 per minute. The patient showed poor nutritional status, weighing 48.7 kg with a height of 169 cm (body mass index [BMI], 17.05 kg/m2). The patient appeared cachectic and exhausted by prolonged admission.
Initial laboratory studies showed pancytopenia with neutropenia (absolute neutrophil count [ANC], 150/╬╝L; reference, >1,500/╬╝L), hyponatremia (131 mmol/L), and increased prothrombin time (12.9 seconds) and partial thromboplastin time (41.1 seconds). C-reactive protein (40.3 mg/L) and erythrocyte sedimentation rate (59 mm/h) were also elevated. Additional routine laboratory test results are shown in Table 1. Screening for infection yielded no evidence of cytomegalovirus infection, or other causes of atypical pneumonia. Cultures of the blood and sputum were negative, but urine culture yielded 1,000 colony-forming units/mL of Gram-positive cocci. On chest X-ray, multiple nodular consolidations were observed in the left upper lung, and a chest computed tomography (CT) scan further demonstrated multiple nodular consolidations in the right upper lobe with a small amount of left pleural effusion and pericardial effusion. No enlarged lymph nodes were identified, and the enlarged spleen measured 21.4 cm.
Initial medications upon admission included aspirin, nicorandil, trimetazidine, bisoprolol, and levothyroxine for the treatment of the underlying conditions. Moreover, total parenteral nutrition was initiated to compensate for the patientŌĆÖs nutritional deficit. Antibiotic therapy was commenced with trimethoprim/sulfamethoxazole, meropenem, and amphotericin B, as was used in the previous clinic. However, as the chest CT scan indicated minimal possibility of active Pneumocystis pneumonia, and polymerase chain reaction for Pneumocystis jirovecii was also negative, amphotericin B was administered as the sole antifungal agent. Additionally, prophylactic antibacterial agents were reduced to cefepime, since the fever had subsided by the time the patient was transferred to Severance Hospital.
Consultations from the departments of hematology, rheumatology, and infection were sought, and the differential diagnosis focused on the evaluation of splenomegaly, pancytopenia, and neutropenia. Peripheral blood smear revealed normochromic, normocytic anemia with a reticulocyte percentage of 0.65%. Screening test results for iron deficiency anemia indicated anemia of chronic disease, with elevated ferritin (3,308.5 ng/mL) and low transferrin (98 mg/dL) along with normal serum iron level (79 ╬╝g/dL). In addition, there was no deficiency of B12 and folate. Bone marrow biopsy revealed an average cellularity of 60%, with no signs of bone marrow failure. The detailed results can be found in Table 2, and follow-up results for complete blood counts are listed in Table 3.
Prior bone marrow biopsies had similarly failed to indicate a diagnosis, which made primary hematologic disease unlikely. Subsequently, investigations were performed for potential autoimmune causes, and these yielded positive anti-nuclear antibodies (ANA) with a titer of 320:1 and rheumatoid factor (RF) levels of 23 IU/mL. After a rheumatology consultation, further studies were performed revealing weakly-positive anti-DNA (19 IU/mL) and anti-cardiolipin immunoglobulin M antibodies (11 MPL-U/mL). Anti-beta2-glycoprotein 1 antibodies and lupus anticoagulant were both negative. Serum C3 was low (36.7 mg/dL), but C4 was normal (11.98 mg/dL). Furthermore, when the patient was evaluated for lupoid manifestations, an oral ulcer was found, but no other autoimmune features were seen, including skin rash, arthritis, muscular weakness, uveitis, and Reynaud phenomenon. Cystatin C was elevated (1.53 mg/L), and cystatin C-based estimated glomerular filtration rate (GFR) was 42.6 mL/min/1.73 m2. Although the creatinine level was normal (0.82 mg/dL), its diagnostic value was limited due to the low BMI of the patient. Considering the ongoing infection, the presence of renal failure was deemed indeterminate. Follow-up investigations showed equivocal levels of anti-DNA antibodies (10 IU/mL), therefore, the elevation of rheumatologic antibodies was tentatively considered to be a by-product of infection upon further consultation.
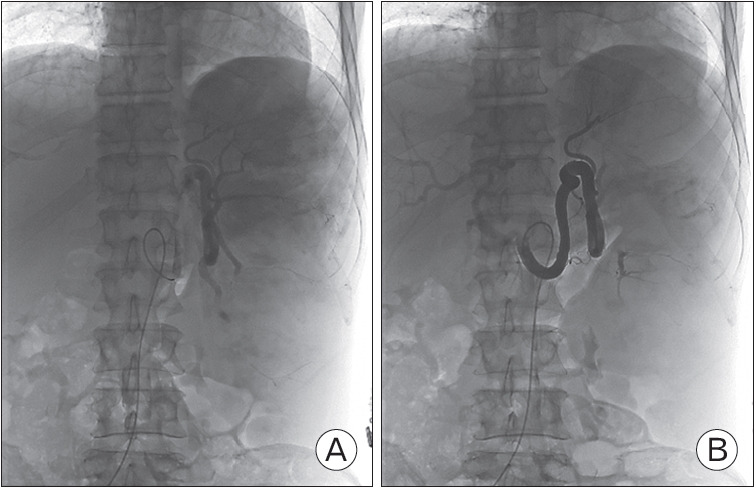
With other possible causes of pancytopenia ruled out, splenomegaly itself was suspected to be the cause of the pancytopenia. During the hospitalization, the pancytopenia progressively worsened, and by day 11 of hospitalization, total white cell count reached 540/╬╝L (neutrophil 35.9%), hemoglobin 6.8 g/dL, and platelet count 57├Ś103/╬╝L. Moreover, onset of sepsis was suspected, with body temperature of 37.0┬░C, blood pressure of 86/52 mm Hg, and pulse rate of 84. After an infusion of 1.5 L of fluid did not raise the blood pressure to baseline levels, filtered red blood cells were transfused, along with norepinephrine at a rate of 4 mL/h. Given the patientŌĆÖs indeterminate diagnosis and deteriorating general condition, splenectomy was considered as a curative treatment option. However, considering the poor condition of the patient, surgical splenectomy was deemed as hazardous, and partial splenic embolization was considered as an alternative. During the course of hospitalization, numerous consultations were sought but due to the failure to establish a definite diagnosis, no specific department could agree to actively transfer the patient from our department. Partial splenic embolization was undertaken on day 17 of hospitalization, using iodixanol as a contrast agent, and ultrasound guidance for arterial access. Vascular access point was right common femoral artery, and after performing celiac angiography and splenic angiography, the lower portion (70%) of the spleen was embolized with gelfoam. Intra-procedural images are shown in Figure 1.
The day after embolization, there were no serious complications and a general increase in blood counts was observed; white blood cell count rose to 820/╬╝L, hemoglobin to 10.0 g/dL, and platelet count was 45├Ś103/╬╝L. However, 2 days after the procedure, at 11 PM, the patientŌĆÖs temperature rose to 38.5┬░C, followed by a rapid drop to 35.7┬░C in an hour accompanied by a decrease in blood pressure to 84/56 mm Hg and a pulse rate increase to 148 per minute. Septic shock was suspected; therefore, norepinephrine and fluids were infused and CT scans of the chest and abdomen were undertaken. Notably, there was no pulmonary embolism or bowel perforation, and the abdominal CT image is shown in Figure 2. After 2 hours of resuscitation, increased rales were heard in the lungs, and oxygen saturation began to fall, eventually reaching 55%. Consequently, mechanical ventilation was initiated. However, given the grim prognosis, the legal guardians of the patient refused to give consent for admission to the intensive care unit for closer monitoring and treatment. The patient began to show signs of cardiac arrest at 5 AM, when cardiopulmonary resuscitation (CPR) was commenced. Nevertheless, spontaneous circulation could not be established and the patient died 60 minutes into CPR at 6 AM.
Written informed consent for publication of this image was obtained from the patient.
DISCUSSION
This case report describes a case of pancytopenia with splenomegaly in an elderly man. Specifically, pancytopenia and accompanying neutropenia (initial ANC 88/╬╝L) were the major issues, and a history of multiple, long-term admissions was severely jeopardizing the patientŌĆÖs quality of life. No definite diagnosis could be made in spite of a range of rheumatologic and hematologic evaluations conducted in our hospital and in previous hospitals. All investigations yielded similar results and did not meet any specific diagnostic criteria. This suggested a diagnostic ŌĆśgray zone,ŌĆÖ where the patientŌĆÖs general condition clearly indicated severe pathologic changes, but no definite diagnosis could be assigned; thus, this case could be described as a case of ICUS. ICUS is defined as an unexplained persistent cytopenia which fails to be diagnosed as myelodysplastic syndrome (MDS) by the accepted criteria. Cytopenia must be substantial and observed for at least 6 months, and upon diagnosis, a thorough hematologic follow-up, similar to that in low-risk MDS is warranted due to its possible progression to MDS or another myeloid disorder over time [3].
The differential diagnoses we considered were aplastic anemia, leukemia, myelofibrosis, and MDS among hematologic causes, and systemic lupus erythematosus (SLE) among autoimmune causes. With respect to hematologic causes, multiple bone marrow biopsies performed at the current and previous hospitals failed to reveal signs of a hypocellular marrow, ineffective hematopoiesis, or myelofibrosis. For rheumatologic causes, the presence of ANA and RF was investigated, and mildly elevated anti-dsDNA antibodies were discovered. Although anti-dsDNA antibodies measured by enzymeŌĆÉlinked immunosorbent assay generally have a high specificity, low or equivocal titers may be induced by conditions other than SLE [4]. Specifically, moderately positive titers indicate 72% specificity and equivocal titers indicate 65% specificity [5]. As the titers never reached strongly positive levels and rather, decreased over time, we presumed that sepsis was responsible for the transient elevation of anti-dsDNA antibodies and rheumatologic factor. As for the presence of renal dysfunction seen in lupus nephritis, both creatinine and cystatin C were measured to estimate the GFR, but the accuracy of creatinine-based estimated GFR was questionable due to the low BMI [6]. Additionally, cystatin C is also known to be affected by positive RF [7]; therefore, the exact renal function status of the patient could not be ascertained.
Splenectomy was initially considered as a curative treatment as splenic sequestration was suspected due to the massive splenomegaly. Furthermore, it has been previously reported that splenectomy may have a curative effect in such cases [1]. However, as the patient was critically ill, partial splenic embolism was performed instead to avoid the risks of surgery [8]. Unfortunately, these measures failed to prevent the eventual death of the patient from septic shock
The role of family medicine is often described as ŌĆśproviding primary care from birth to the end of life.ŌĆÖ In the present case, we found ourselves in a dilemma regarding the optimal management of the patient. This case represented a diagnostic ŌĆśgray zone,ŌĆÖ where the referral was rejected while no definitive treatment options could be decided upon even after consultations, due to the uncertain diagnosis. However, the condition was having a severely detrimental impact on the quality of life of the patient with no notable improvement over the long clinical course; therefore, the desire of the patient and his family for definitive treatment was considered before deciding the management plan. With consultations regarding medically unexplained physical symptoms becoming more common in recent times, similar dilemmas may occur in such ŌĆśgray zoneŌĆÖ patients. Currently, although all subspecialties put significant emphasis on disease management, less attention may be paid to the desires of the patients and their families. Family medicine may fill in the gaps here. As providers of comprehensive care, we can optimally incorporate the personal values of the patients and their families into the medical decisions, and thus help in deciding the best course of action in similar situations. We hope that our experience in this case may give some insight into the management of future patients in the ŌĆśgray zone.