INTRODUCTION
With increasing age, the number of patients visiting primary health care facilities due to memory decline has been increasing. The spectrum of complaints of memory decline ranges from subjective memory impairment (SMI) to mild cognitive impairment (MCI) to dementia. The clinical significance of SMI and amnestic MCI has been growing, as these conditions may represent a transitional state between normal cognition and Alzheimer’s disease (AD) [1,2]. However, not all patients progress to AD, and early detection of individuals who are at high risk of AD is important to predict prognosis and reduce social burdens.
The hippocampus is the earliest affected and most vulnerable structure in AD [3], and medial temporal atrophy (MTA) is known as the hallmark of AD. Brain magnetic resonance imaging (MRI) is, therefore, a useful screening tool to detect MTA with a predictive value for AD. However, hippocampal atrophy can also be observed in various neurodegenerative diseases, such as vascular dementia, dementia with Lewy bodies, and Parkinson’s disease [4,5]. It is necessary to consider whether or not MTA is in fact a specific marker of AD.
Indeed, several previous studies have reported inconsistent results regarding the value of MTA for detecting the prodromal stage of AD [6,7] and for differentiating AD from other dementias [8]. However, these studies are limited by the pathologically unproven AD diagnosis; to the best of our knowledge, in most of these studies, AD was diagnosed based on clinical criteria without any evidence of β-amyloid deposition from pathological data or on positron emission tomography (PET) scans [8-10]. Therefore, the aim of this study was to investigate the value of MTA for detecting 18F-florbetaben (18F-FBB) PET-proven AD pathology. If MTA can reflect underlying AD pathology, a brain imaging study can be actively recommended as a screening tool for dementia in primary care.
METHODS
1. Subjects
We consecutively enrolled 265 patients complaining of cognitive decline at the dementia outpatient clinic, Severance Hospital (Yonsei University College of Medicine), from March 2015 to December 2017. All subjects underwent a detailed neuropsychological assessment (Seoul Neuropsychological Screening Battery [SNSB]) to assess their level of cognitive performance [11]. To uncover the underlying disease that caused the cognitive decline, all subjects underwent brain MRI, 18F-fludeoxyglucose (18F-FDG) PET, and 18F-FBB PET. Apolipoprotein E (APOE) genotyping was performed in 260 patients (five patients declined to be genotyped). All subjects also underwent a neurologic examination, and 18F-fluorinated N-3-fluoropropyl-2-beta-carboxymethoxy-3-beta-(4-iodophenyl) nortropane (18F-FP-CIT) PET scans were performed in 132 subjects who showed parkinsonism. 18F-FDG PET, 18F-FBB PET, and 18F-FP-CIT PET revealed regional hyper-/hypometabolism, β-amyloid deposition, and nigrostriatal dopaminergic degeneration, respectively. All these diagnostic work-up procedures were carried out within 6 months, and clinical diagnoses were performed based on the clinical features and neuroimaging findings. This study was approved by the Yonsei University Severance Hospital institutional review board (IRB approval no., 4-2016-0210). The need for informed consent was waived because of the retrospective nature of the study.
2. Neuropsychological Assessment
The SNSB covers five cognitive domains: attention and working memory (forward/backward digit span task and letter cancellation test); language and related functions (the Korean version of the Boston Naming Test, calculation, and praxis); visuospatial function (the Rey Complex Figure Test [RCFT] copy and interlocking pentagon); verbal and visual memory (immediate recall/delayed recall/recognition test using the Seoul Verbal Learning Test [SVLT] for verbal memory; immediate recall/delayed recall/recognition test using the RCFT for visual memory); and frontal/executive function (contrasting program, go/no-go test, the Controlled Oral Word Association Test, and the Stroop test). In addition, the Korean version of the Mini-Mental State Examination (K-MMSE) was used to assess general cognition. We used z-scores to assess the level of cognitive performance. A z-score was defined as where the score was positioned in the distribution of scores for age- and education-matched normal subjects. The memory composite scores were calculated by dividing the sum of the z-scores by the number of tests in the memory function domain.
3. Visual Rating of Medial Temporal Atrophy and White Matter Hyperintensities
1) Brain MRI acquisition
MRI scans were acquired using a Philips 3.0 T scanner (Philips Achieva; Philips Medical Systema, Best, The Netherlands) with a SENSE head coil (SENSE factor=2).
2) Visual rating of MTA
The Scheltens scale was used to rate MTA [12]. According to this scale, visual assessment of MTA is scored on a scale of 0 (no atrophy) to 4 (end-stage atrophy). The characteristics of each group were defined as the width of the choroid fissure, the width of the temporal horn, and the height of the hippocampal formation (Table 1). Then, we used four criteria to assess the extent of MTA in each subject. According to criterion 1, the MTA score was calculated by averaging the right and left MTA scores for each subject as a continuous variable. For all other criteria, the MTA findings were classified into normal or abnormal according to the following cut-off scores: criterion 2: a cut-off score of 1.5 was used (i.e., an average MTA score equal to or greater than 1.5 was regarded as abnormal) [13]; criterion 3: an age-dependent cut-off score (i.e., 3 for subjects ≥75 years old, and 2 for subjects <75 years old, in either hemisphere) was used [14]; and criterion 4, age was further subdivided using cut-off scores of 1, 1.5, and 2.0 for subjects <65, 65–74, and >75 years old, respectively [15].
3) Visual rating of WMHs
The modified Fazekas scale was used to describe the extent of periventricular and deep white matter hyperintensities (WMHs) [16]. Periventricular WMH was classified as P1 (cap and band <5 mm), P2 (5 mm ≤ cap or band <10 mm), or P3 (10 mm ≤ cap or band) and deep WMH was classified as D1 (maximum diameter of deep white matter lesion <10 mm), D2 (10 mm ≤ lesion <25 mm), or D3 (≥25 mm). We subdivided the subjects into three groups: the minimal ischemia group (D1P1 and D1P2), the moderate ischemia group (D1P3, D2P1, D2P2, and D2P3), and the severe ischemia group (D3P1, D3P2, and D3P3) [17].
4. Visual Assessment of Amyloid Deposition
1) 18F-FBB PET data acquisition
18F-FBB PET scans were performed using Discovery 600 (General Electric Healthcare, Milwaukee, MI, USA). The images were acquired with a 256×256 matrix 90 minutes after administration of 300 MBq (8 mCi) FBB for 20 minutes. Then the images were reconstructed with an ordered-subsets expectation maximization algorithm in an iso-0.98-mm voxel size.
2) Visual assessment of 18F-FBB PET images
The regional cortical tracer uptake (RCTU) and brain β-amyloid plaque load (BAPL) scoring systems were used to visually assess the deposition of β-amyloid [18]. The RCTU scoring system grades the tracer uptake in the lateral temporal cortex, frontal cortex, posterior cingulate cortex/precuneus, and parietal cortex as follows: grade 1, no binding; grade 2, minor binding; grade 3, pronounced binding. Based on the RCTU scores, the BAPL scoring system classifies the results into β-amyloid-negative PET scans (BAPL score 1) and β-amyloid-positive PET scans (BAPL scores 2 and 3).
6. Statistical Analyses
Multivariable logistic regression analyses were performed to investigate whether the MTA and K-MMSE z-scores, which can be easily obtained in primary health care clinics, were predictive of underlying AD pathology (model 1). In addition, we performed multivariable logistic regression analyses by entering additional variables including age, sex, years of education, WMH, APOE genotype, and memory composite scores in various combinations. Model 2 included age, sex, years of education, WMH, MMSE z-scores, and MTA as variables. Model 3 included age, sex, years of education, WMH, APOE genotype, memory composite scores, and MTA as variables. The discriminatory power of variables was investigated by receiver operating characteristic analyses. A Bootstrap method was used to compare the area under the curve (AUC) between the variables. The statistical analyses were performed with IBM SPSS software ver. 23.0 (IBM Corp., Armonk, NY, USA) and R software package ver. 3.4.0 (http://www.r-project.org), and a two-tailed P<0.05 was considered significant.
RESULTS
1. Demographic Characteristics
Of the 265 patients, 121 were diagnosed with AD-related cognitive impairment (98 with pure AD pathology and 23 with mixed pathology). The other 144 were diagnosed with Lewy bodies-related cognitive impairment (i.e., Parkinson’s disease or dementia with Lewy bodies; n=42), vascular cognitive impairment (n=32), other pathologies (i.e., epileptic cognitive impairment, tauopathy, normal pressure hydrocephalus and others; n=23), and undetermined pathologies (n=47). There were no significant differences in age, sex, education, K-MMSE scores, and WMHs between the AD and non-AD pathology groups. The prevalence of the APOE ε4 allele and dementia were higher in patients with AD pathology. In addition, the patients with AD-related cognitive impairment had lower memory composite scores and tended to show more severe MTA than those with non-AD pathology (Table 2).
2. Association between Medial Temporal Atrophy and Alzheimer’s Disease Pathology
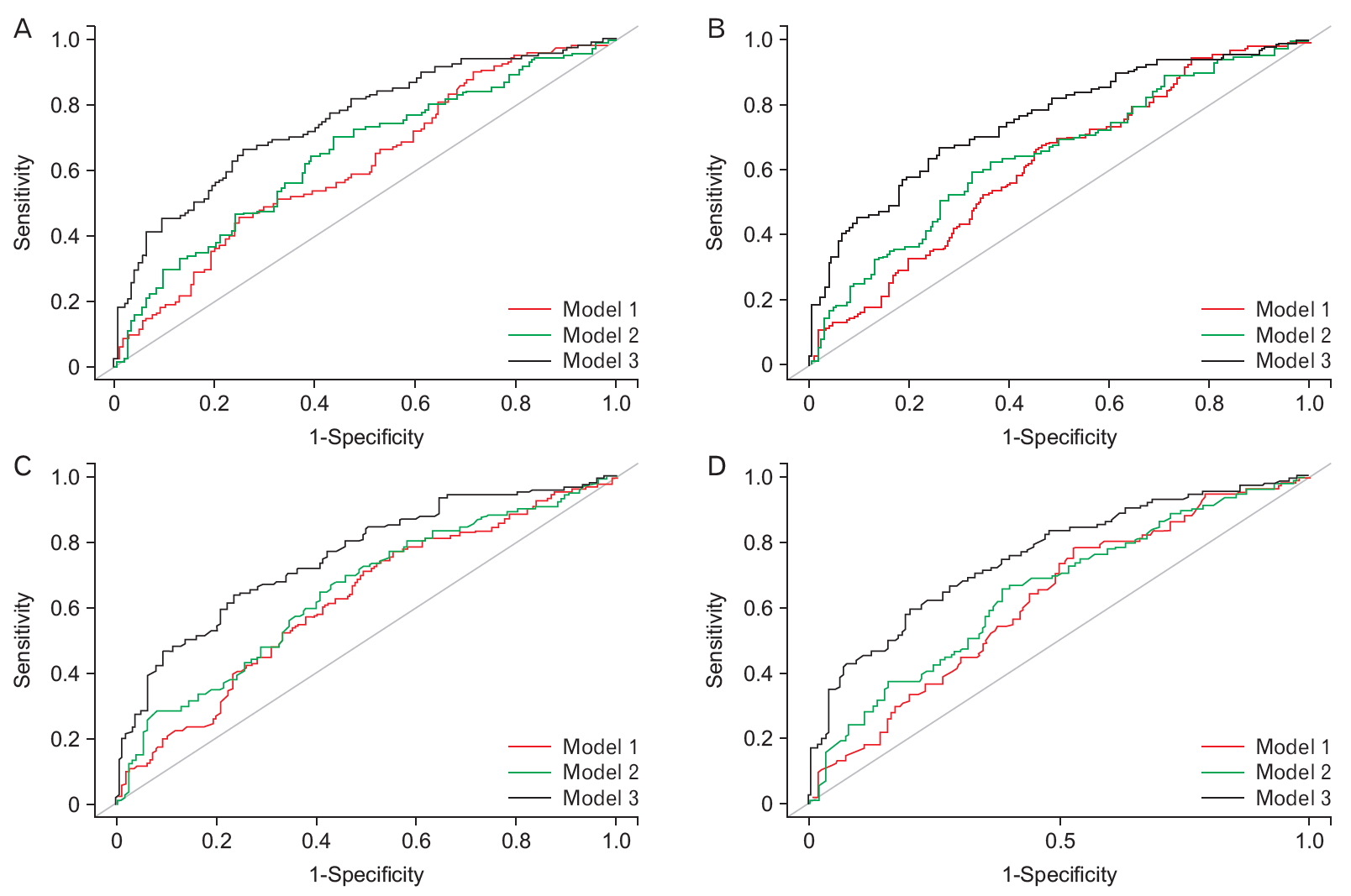
Multivariable logistic regression analyses were performed using the MTA and K-MMSE z-scores as variables (model 1). The results show that MTA was not an independent predictor for the presence of AD pathology in patients with cognitive complaints (MTA criterion 1, P=0.222; criterion 2, P=0.302; criterion 3, P=0.422; criterion 4, P=0.771) (Table 3). The analyses were not powerful enough to differentiate AD-related cognitive impairment from non-AD-related cognitive impairment; the AUCs were 0.616 (95% confidence interval [CI], 0.549–0.684), 0.617 (95% CI, 0.549–0.683), 0.618 (95% CI, 0.551–0.686), and 0.618 (95% CI, 0.551–0.686) for the MTA criteria 1, 2, 3, and 4, respectively (Table 4, Figure 1).
3. Receiver Operating Characteristic Analyses for Differentiating Alzheimer’s Disease from Non-Alzheimer’s Disease
Additionally, we performed multivariable logistic regression analyses by entering additional variables. Model 2 showed again that MTA was not an independent predictor for the presence of AD pathology in patients with cognitive decline (MTA criterion 1, P=0.202; criterion 2, P=0.352; criterion 3, P=0.383; criterion 4, P=0.831) (Table 3). This statistical model did not have enough discriminatory power to discriminate between AD- and non-AD-related cognitive impairment either (AUC, 0.647; 95% CI, 0.580–0.713) for MTA criterion 1, (AUC, 0.644; 95% CI, 0.578–0.711) for MTA criterion 2, (AUC, 0.644; 95% CI, 0.578–0.711) for MTA criterion 3, and (AUC, 0.645; 95% CI, 0.578–0.711) for MTA criterion 4 (Table 4, Figure 1).
Model 3 also showed that MTA did not significantly predict the presence of AD pathology (MTA criterion 1, P=0.686; criterion 2, P=0.732; criterion 3, P=0.524; criterion 4, P=0.781) (Table 3). However, this model had a fair discriminatory power with AUCs of 0.751 (95% CI, 0.692–0.810), 0.752 (95% CI, 0.694–0.811), 0.751 (95% CI, 0.692–0.810), and 0.751 (95% CI, 0.692–0.809) for MTA criteria 1, 2, 3, and 4, respectively (Table 4, Figure 1). The AUCs for model 3 were significantly greater than those for the other models (e.g., for the MTA criterion 4: model 1 versus model 2, P=0.963; model 1 versus model 3, P=0.001; model 2 versus model 3, P=0.003) (Table 4).
DISCUSSION
The present study investigated whether MTA observed on brain MRI scans accurately reflects the underlying AD pathology confirmed by 18F-FDG PET and 18F-FBB PET scans. The major findings of the study were as follows: (1) in the multiple logistic regression analyses, MTA was not an independent predictor of underlying AD pathology; and (2) the predictive power of the statistical model significantly increased (model 3) when multiple variables including age, sex, years of education, WMH, APOE genotype, and memory composite scores were considered together. However, the statistical model (model 2) including age, sex, years of education, WMH, and K-MMSE z-scores as variables did not predict the underlying AD pathology effectively. These findings suggest that K-MMSE and brain imaging studies, which are mainly used for screening of dementia in primary health care facilities, have a weak predictive power on whether or not the patient’s memory decline is caused by AD-related cognitive impairment.
Dementia is a clinically observable outcome of the cumulative burden of multiple pathological insults in the brain and can result from various neurodegenerative disorders (e.g., AD, Parkinson’s disease, dementia with Lewy bodies, frontotemporal dementia, and others) as well as non-neurodegenerative conditions (e.g., vascular cognitive impairment and vitamin deficiency). It is important to uncover the underlying pathogenesis leading to cognitive decline because the prognosis and therapeutic approach depend on the specific condition. The most common pathological condition underlying dementia is AD [24], and many patients who visit the outpatient clinic due to cognitive decline would thus like to know if their problems are caused by AD-related cognitive impairment. MTA is known as one of the typical features of AD [3], and brain MRI scans are, thus, commonly used to reveal the underlying AD pathology. In fact, a number of studies on the possibility of predicting AD by MRI have been conducted [6-10], but the results are still controversial.
The present study demonstrates that the visual rating of MTA on brain MRI scans do not provide crucial information in predicting underlying AD pathology. This is probably due to the following reasons. (1) MTA is not a specific finding of AD. Previous studies have suggested that hippocampal atrophy is also commonly found in healthy elderly people [4], particularly in those of advanced age, as well as in patients with frontotemporal dementia [25], dementia with Lewy bodies, and Parkinson’s disease [5]. (2) The structural abnormalities found on brain MRI may be minimal to mild in the preclinical stage of AD or amnestic MCI. According to Alzheimer’s pathological cascade [26], β-amyloid plaque deposition precedes clinical symptoms and reaches a plateau by the time clinical symptoms appear. Then, tau-mediated neuronal damage and dysfunction occur later, and correlate well with clinical symptom severity. Structural abnormalities assessed by brain MRI is the last biomarker, and the brain atrophy rate accelerates as patients approach dementia. In addition, according to Braak and Braak [27], the AD-related neurofibrillary changes occur in six stages: transentorhinal stages (stages 1 and 2), limbic stages (stages 3 and 4), and neocortical stages (stages 5 and 6). In the transentorhinal stages, the neurodegenerative changes in the brain remain below the threshold, which indicates clinical symptoms. The characteristic brain lesions, such as destruction of limbic circuits, become evident in the limbic stage. In this regard, structural alterations observed on brain MRI alone may be a less sensitive tool for detecting underlying AD pathology in patients with either SMI or amnestic MCI.
Model 2, which included age, sex, years of education, and WMH as additional variables, did not have enough discriminatory power to predict the underlying AD pathology, while model 3, which additionally included APOE genotype and memory composite scores, predicted the underlying AD pathology, regardless of the MTA criteria. The APOE ε4 allele is a well-established generic risk factor for late-onset AD through both β-amyloid-dependent and β-amyloid-independent pathways [28]. The prominent memory dysfunction is the pathognomonic neuropsychological profile of AD-related cognitive impairment, given that early AD pathology frequently affects the neuroanatomical networks for episodic memory before other networks [29]. Therefore, at least a detailed memory function test (i.e., the SVLT and RCFT in the SNSB) should be considered in patients who complain of memory decline, to differentiate AD-related cognitive impairment from non-AD-related cognitive impairment.
Our study has some limitations. First, selection bias may have occurred when we enrolled the patients with non-AD-related cognitive impairment. We retrospectively recruited patients who underwent both 18F-FDG PET and 18F-FBB PET to exclude the possibility of underlying AD pathology. These subjects may be more likely to have MTA and thus undergo 18F-FBB PET, which might have led to the results showing that MTA was not an independent predictor for the presence of AD pathology. Second, this study was based on visual ratings of MTA and 18F-FBB uptake, which may be somewhat subjective. An objective measurement or quantification of medial temporal volume and β-amyloid deposition is needed to confirm our findings in the future. Third, although MRI is the preferred imaging modality in the diagnostic work-up of cognitive decline, MRI is not available to a number of patients in primary health care clinics. Instead, brain computed tomography can be used as an alternative for MRI, which has been reported to yield reliable information on MTA [30].
In conclusion, our results suggest that the visual rating of MTA alone is insufficient to accurately predict the presence of AD pathology. In order to determine whether the patient is at high risk of AD or not, it is necessary to comprehensively consider various other factors such as APOE genotype and the results of a detailed memory function test.