INTRODUCTION
Myelodysplastic syndromes (MDS) are a heterogeneous group of acquired clonal stem cell disorders that cause ineffective and dysplastic production of blood cells and can transform to acute myeloid leukemia (AML). MDS is most common in older age groups and is probably the most common hematological malignancy in adults 60 years of age and older in the United States, affecting about 1 in 500 people in this age group.1) About 15,000 new cases of MDS are diagnosed annually in the United States, and with the number of people over the age of 65 expected to double by 2030, this number is expected to increase.2) It is important for primary care physicians (PCPs) to recognize the signs and symptoms of MDS because they are often patients' first point of contact in the health care system. PCPs therefore play an important role in ensuring early referral and diagnosis of MDS patients. There is also a shortage of oncologists which increases the importance of the role of PCPs in care of MDS patients.3) It has been shown that more than 90% of PCPs co-manage comorbid conditions, evaluate and treat depression, establish do not resuscitate, refer to hospice, and prescribe opiates in cancer patient management, though they are generally less involved in treatment decision making.4) In this article, we will review the common manifestations, diagnostic work-up, and management of MDS for PCPs, with a focus on when to suspect MDS, when a referral is needed, and how to provide appropriate supportive care for patients diagnosed with MDS.
ETIOLOGY
MDS can be either primary (de novo) or secondary to previous treatment with chemotherapy, radiation therapy, or immunosuppressive medications. Environmental and occupational factors such as exposure to ionizing radiation, agricultural chemicals such as pesticides, or industrial solvents such as benzene, are associated with increased MDS risk.2) The cities of Hiroshima and Nagasaki, Japan report higher than average rates of MDS, especially among those exposed to radiation from the atomic bombs when they were young.5) Cigarette smoking, which is the greatest non occupational source of benzene exposure, has also been associated with greater MDS risk in several studies.6)
Secondary, or therapy-related, MDS (t-MDS) accounts for about 10% of all MDS cases and has a worse prognosis than de novo MDS.2) It typically occurs after chemotherapy or radiation treatment for a previous cancer, or following prolonged use of immunosuppressive drugs in patients with autoimmune disorders or organ transplantation. Among MDS patients with one or more primary tumors, the most common types of primary tumor include non-Hodgkin or Hodgkin lymphoma, acute lymphoblastic lymphoma, and cancers of the breast, prostate, urinary system, and lung or bronchus.7,8) The median time from diagnosis of the primary tumor to the diagnosis of MDS is 4.5 years.7)
Two of the most common and best understood forms of t-MDS occur after treatment with alkylating agents and topoisomerase II inhibitors. T-MDS following treatment with alkylating agents typically presents as MDS with monosomy 7, del(5q), or monosomy 5, after a latency period of about 5 to 7 years. T-MDS is also occasionally found after treatment with topoisomerase II inhibitors, but more commonly the patient presents with full-blown AML after a latency period of just 1 to 3 years. Topoisomerase II inhibitors are associated with translocations involving chromosome bands 11q23 or 21q22.9)
Based on these risk factors, PCPs should have a high index of suspicion for MDS in patients treated with chemotherapy or radiation for a prior cancer, patients with a history of autoimmune disorders, organ transplantation, and immunosuppressive therapy, and patients with potential for significant radiation or chemical exposure due to environmental, occupational, or lifestyle factors.
EPIDEMIOLOGY
Both de novo and secondary MDS are more common among older patients. In the United States, the incidence rate increases from about 0.5 per 100,000 per year among individuals younger than 50 years to 89 per 100,000 individuals older than 80 years.10) Patients with several congenital conditions, including Fanconi anemia, Down syndrome, and Bloom syndrome, tend to present at younger ages than the general population.2) For example, a study of data from the International Fanconi Anemia Registry found that the actuarial risk of developing MDS or AML for individuals with Fanconi anemia was 52% by 40 years of age.11)
MDS is more common in men than women.7) Among different racial groups in the United States, Caucasians have the highest incidence, while American Indians and Alaska Natives have the lowest.7) Notably, some regions of East Asia tend to report cases of MDS at significantly younger ages than the rest of the world. A study of 508 patients in China found a median age at diagnosis of 49, compared to the United States median of 65 to 73,12) while a smaller study in Korea found a median age of 57.13) Both studies also reported different clinical profiles than those of a typical Western patient, and suggested a combination of genetic and environmental factors that likely account for the differences. In the absence of other known risk factors, PCPs should have the highest index of suspicion for MDS in older, Caucasian men.
CLINICAL PRESENTATION
Patients with MDS can be asymptomatic, and their disease may be diagnosed only after incidental finding of cytopenias in a routine blood test. The most common cytopenia associated with MDS is anemia, which is found in about 80% to 85% of patients.14) Anemia associated with MDS is usually macrocytic (Figure 1) or normocytic.14) Most symptomatic patients display classic symptoms and signs of anemia, such as pallor, chronic fatigue, shortness of breath, cognitive impairment, and chest pain or palpitations that are exacerbated by exertion. Thrombocytopenia is found in about 35% to 45% of patients, and neutropenia in about 40%.14) Patients with thrombocytopenia may complain of easy bruising or frequent minor mucocutaneous bleeding and may display purpura or petechiae. In more extreme cases, increased risk of gastrointestinal bleeding or intracranial hemorrhage may occur. Fatigue and frequent bacterial infections, especially of the skin, are common complaints of patients with neutropenia. Bone marrow aspiration and biopsy in MDS patients typically reveal hypercellular bone marrow, but about 5% to 10%, most commonly women, present with hypocellular bone marrow.15) PCPs should consider MDS in elderly patients with persistent macrocytic anemia, multiple cytopenias, or dysplastic peripheral blood cells, after exclusion of other conditions.
APPROACH TO DIAGNOSIS OF MDS IN PRIMARY CARE SETTINGS
MDS can be difficult to diagnose due to the high rate of co morbidities and other health concerns related to advanced age, some of which can mimic the presentations of MDS. For example, anemia is common among the elderly and is often unrelated to any serious hematological problem. One study of patients aged 65 years or older found that more than 10% had some form of anemia, with causes ranging from minor nutrient deficiencies to major medical disorders.16)
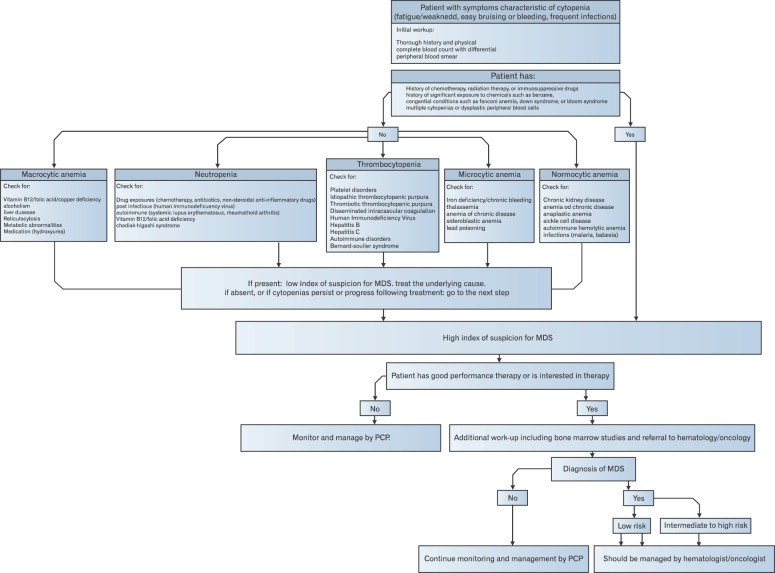
Since PCPs are often the first contact point for patients in the health care system, a PCP who suspects the possibility of MDS should implement the following diagnostic approach to rule out other possible causes of the patient's clinical presentation and cytopenias (Figure 2). 1) A detailed history, with a focus on occupation, medications, exposures, and alcohol abuse. 2) A physical examination, checking for pallor, purpura, petechiae, hepatomegaly, splenomegaly, and lymphadenopathy. Organomegaly is not common in MDS; however, it can be seen, particularly among patients with overlapping features of MDS and chronic myeloproliferative disorders. 3) A complete blood cell count with differential and reticulocyte count, as well as a peripheral blood smear. The presence of circulating blasts in a peripheral smear mandates urgent hematology-oncology referral. 4) Liver, renal, and thyroid function tests. 5) Screening for nutrient deficiencies. If the patient presents with macrocytic anemia, the most important deficiencies to check for include vitamin B12, folate, and copper. Also determine if the patient is taking a zinc preparation, since zinc can decrease serum copper levels.17) Copper deficiency can be associated with cytopenias and dysplastic morphological changes in blood cells. For microcytic anemia, conduct iron studies and screen for lead poisoning, thalassemias, and sideroblastic anemia. 6) Screening for chronic blood loss, including fecal occult blood testing. 7) Screening for relevant infections and autoimmune conditions, such as human immunodeficiency virus, hepatitis B and C, malaria, babesiosis, autoimmune hemolytic anemia, idiopathic thrombocytopenic purpura, rheumatoid arthritis, and systemic lupus erythematosus.
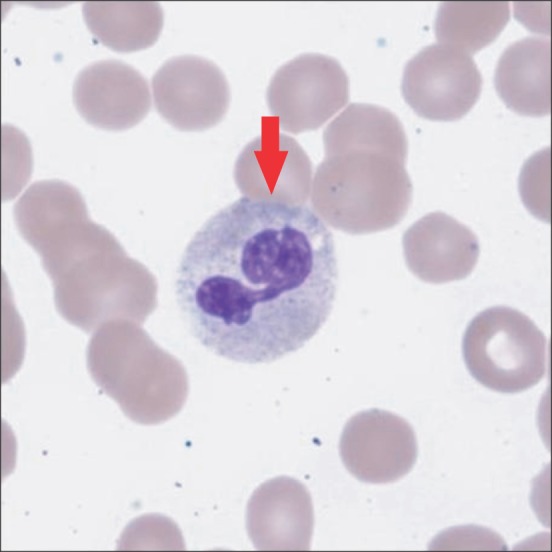
Diagnosis of MDS can be challenging, particularly in isolated cytopenias. It requires performing a bone marrow aspiration and biopsy, but as avoiding bone marrow biopsy or hematology referral when it is not needed can save resources, it would be reasonable for PCPs to closely monitor elderly patients with chronic mild cytopenias who are asymptomatic, who do not want therapeutic intervention, or who would not benefit from chemotherapy due to co-morbidities, rather than referring the patient to a specialist for a more detailed and invasive work-up. On the other hand, bone marrow aspiration and biopsy or hematology referral will be indicated for symptomatic patients, patients requiring blood transfusions, patients with isolated cytopenia with dysplastic morphology (Figures 3, 4) or circulating blasts in peripheral blood, and patients with progressive single or multiple cytopenias.
Cytogenetic abnormalities are detected in about 45% to 50% of MDS patients14) and the specific abnormality found can have important implications for prognosis and treatment. The most common abnormalities include loss of the long arm of chromosomes 5, 7, and 20, loss of chromosome 7, and trisomy 8.15) Identifying MDS with isolated del(5q) is especially important due to the relative ease of treatment with lenalidomide.18) In addition to its characteristic karyotypic abnormality, MDS with del(5q) is associated with a clinical presentation of macrocytic anemia, normal or slightly elevated platelet counts, hypolobated megakaryocytes, and less than 5% bone marrow blasts.19)
To assist in the diagnostic process, a set of minimum diagnostic criteria has been established for MDS. In order to establish a diagnosis of MDS, two 'prerequisite' criteria must be fulfilled, as well as at least one out of three possible decisive criteria.20)
The diagnostic prerequisites for MDS are marked and constant cytopenia lasting six months or more (unless cytogenetic studies reveal MDS earlier) in at least one hematopoietic cell lineage and exclusion of other conditions as the primary reason for cytopenia and/or dysplasia.20) Additionally, at least one of the following decisive criteria must be met: 1) dysplasia in at least 10% of all cells in one or more cell lineages (bone marrow aspirate), or more than 15% ringed sideroblasts, which can be seen only with Prussian blue staining; 2) one or more cytogenetic abnormalities characteristic of MDS; 3) a constant blast cell count of 5% to 19% in bone marrow.20)
PROGNOSIS AND TREATMENT
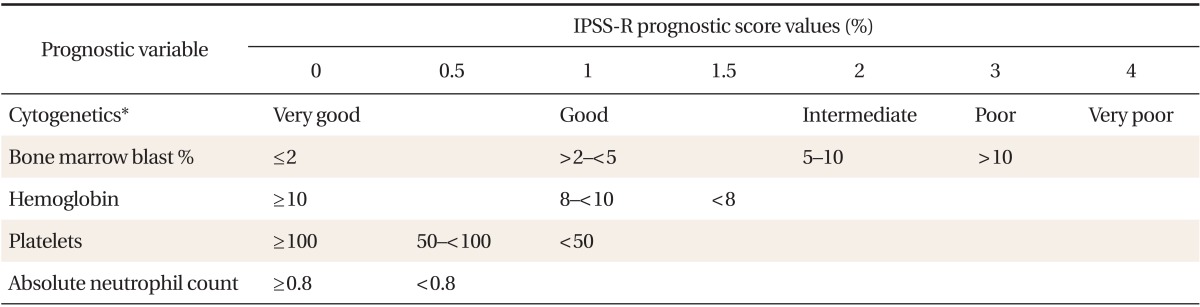
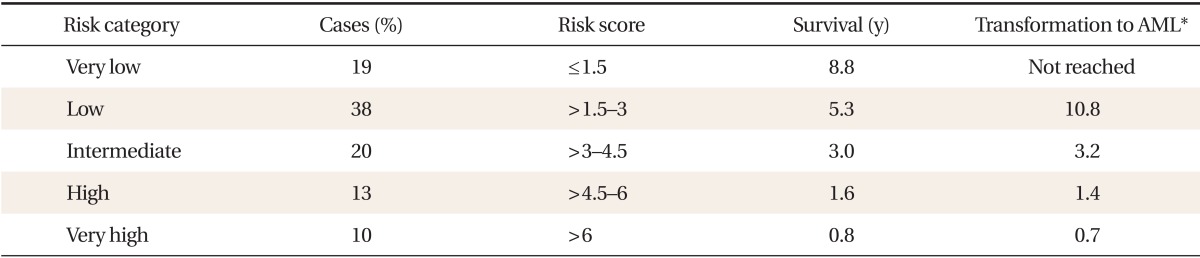
There are several systems that are used for prognostication of MDS. One of the most commonly used is the Revised International Prognostic Scoring System (IPSS-R), which takes into consideration the percentage of blast cells, the karyotype, and the number of blood cell lineages involved (Tables 1, 2). The higher the percentage of bone marrow blasts and the number of cell lines involved, the worse the prognosis is. A low-risk MDS patient has < 5% blasts, good karyotype, and isolated cell line involvement. Patients with lower-risk MDS typically survive 3.0 to 8.8 years, while those with higher-risk MDS typically develop AML or die of complications of MDS (usually bleeding or infection) within 1.5 years.21)
In most cases, the treatment goals for patients with MDS will be to prevent or delay the transformation to AML and to improve the patient's quality of life. The only proven curative therapeutic modality is allogeneic hematopoietic stem cell transplant (allo-SCT). About 35% to 40% of patients who undergo this treatment experience a long-term remission.16) Unfortunately, the rate of transplant-related mortality remains high and many patients are not eligible for the treatment due to advanced age and/or the presence of co-morbidities.14) As a result, allo-SCT is typically performed in less than 5% of patients.22) Better outcomes of allo-SCT are associated with lower IPSS score, younger age, lower blast percentage, and lower-risk cytogenetics.17) For high- and intermediate-risk patients, a study found that those who received transplants soon after diagnosis got the most benefit.18)
Treatment modalities for patients who are not eligible for allo-SCT are tailored based on the risk category, age, and performance status. Often these patients are treated with chemotherapy such as hypomethylating agents (azacitidine and decitabine) or cytotoxic chemotherapy. These treatments do not cure MDS, but they prolong the patient's life and delay the transformation to AML. A multicenter phase III trial comparing azacitidine (75 mg/m2 subcutaneously for 7 days every 28 days) to supportive care found that the median time to death or transformation to AML was 21 months in patients treated with azacitidine, compared to 12 months in patients receiving only supportive care.15) Other common therapeutic interventions for patients with low- to intermediate risk categories include immunosuppressive therapy with antithymocyte globulin and cyclosporine for patients with hypoplastic MDS, and lenalidomide for patients with del(5q) chromosomal abnormality. A phase II study of lenalidomide found that 67% of 148 patients with del(5q) and transfusion-dependent anemia achieved transfusion-independence within 24 weeks, and an additional 9% reduced the frequency of required transfusions by 50% or more.23) Experimental therapies in clinical trials may also be a treatment option for selected patients.
Almost all MDS patients will require red cell or platelet transfusions at some point. One study found that about 65% of MDS patients had received a blood transfusion since diagnosis and about 52% had received a transfusion in the past three months.1) For some patients, the need for transfusions will be only occasional, but about 22% of low-risk and 68% of high-risk patients are transfusion-dependent.1) Erythropoiesis-stimulating agents, most commonly recombinant erythropoietin, should be considered for lower-risk patients (excluding the del(5q) MDS subgroup) with hemoglobin levels of 10 g/dL or lower, especially if pretransfusion erythropoetine levels are below 500 IU/L.18) This can reduce the necessity for packed red blood cell (PRBC) transfusions through stimulation of the patient's own red blood cell production. PRBC transfusions are typically recommended for hemoglobin levels less than 8 g/dL, but there is no single hemoglobin threshold and the trigger level should be adjusted based on the patient's clinical presentation, especially in patients with underlying cardiopulmonary conditions.24) Iron chelation therapy is recommended for low-risk patients with life expectancy of more than one year who have received more than 20 to 30 units of PRBC.17) Serum ferritin levels of 1,000 g/L are typically recommended as the threshold for starting iron chelation therapy.25)
Neutropenia and thrombocytopenia can also result in serious complications in patients with MDS. Granulocyte colony-stimulating factor injections can help keep neutrophil counts above 1 ├Ś 109/L for severely neutropenic patients.18,26) Prophylactic antibiotics have no proven role in patients with MDS and are not recommended for patients with neutropenia related to MDS.2) However, neutropenic fever in MDS patients should be regarded as a medical emergency, requiring immediate administration of empiric broad-spectrum antibiotics and often hospitalization.2) For thrombocytopenic patients, platelet transfusions are typically indicated when platelet levels drop to less than 10,000 plts/┬ĄL.27) The use of androgens such as Danazol can also improve thrombocytopenia.18)
ROLE OF PCPS IN MANAGEMENT OF MDS
PCPs are actively involved in care of patients with cancer, particularly in an era of increasing number of cancer patients and a shortage of oncologists.28) In the face of limited resources, PCP can improve access and quality of care in patients with MDS. Patients with lower-risk MDS may follow up primarily with their PCP for several years once the initial diagnosis and treatment plan has been developed, but it is important that patients with higher-risk MDS are referred to a hematologist/oncologist, due to the higher-risk of AML transformation or mortality from complications of cytopenias within 1.5 years of diagnosis.2) PCPs, in collaboration with hematologists/oncologists, can play an important role in monitoring the patient for signs of infections, bleeding, and severe anemia that may lead to a rapid deterioration in the patient's status, as well as tracking the patient's general quality of life both on and off therapy. It has been shown that communication between PCPs and cancer specialists is lacking and having a plan such as a shared-care model or guidance with something such as a Survivorship Care Plan would likely enhance the ability of the PCP to contribute appropriately.28) This communication is critical as the patient themselves will not likely know who should be responsible for what care and this can lead to confusion and frustration for the patient.29)
PCPs, especially those who have established, long-term professional relationships with their patients, can also assist with counseling. Regardless of treatment, it is important to encourage realistic expectations in higher-risk patients in particular, and provide supportive end-of-life counseling as necessary.
CONCLUSION
MDS is probably the most common hematologic malignancy in the elderly and the number of cases is expected to rise as the average age of the population increases, so PCPs will encounter patients with MDS or suspected MDS on a more frequent basis. Becoming familiar with the basic principles of MDS diagnosis and management can help PCPs ensure earlier diagnosis and better care for patients affected by MDS, improving patients' quality and longevity of life particularly when there are increasing numbers of MDS patients. In the face of limited resources PCPs can have an important role in improving access, coordinating cancer care and optimizing quality of care in patients with MDS.